

Our body consists of different types of cells and the cell-to-cell heterogeneity is characterized by combinations of different ribonucleic acids (RNAs). Information about types and amounts of RNAs in cells helps us understand states and functions of cells, tissues, and organs.
Single-cell RNA sequencing (scRNA-seq) methods measure types and amounts of RNAs in individual cells. However, conventional scRNA-seq methods cannot detect non-poly(A) RNAs, a class of RNAs which are involved in normal development and diseases, and cannot provide full-length coverage of RNAs. Therefore, development of a novel scRNA-seq method is needed to gain full information on RNAs in single cells.
As described in Nature Communications, Tetsutaro Hayashi, Haruka Ozaki and their co-authors used BioJulia to deeply analyze data of their newly-developed single-cell RNA sequencing (scRNA-seq) method, RamDA-seq, which can measure poly(A) and non-poly(A) RNAs and provide full-length coverage.
The goal of this research is to help scientists study cell differentiation and organ regeneration and facilitate development of diagnostic markers for transplanted cells and rare cell populations.
According to Dr. Ozaki, a bioinformatics scientist in the team, “In developing scRNA-seq methods, it is important to evaluate whether and what RNAs in single cells are successfully measured. The raw data from scRNA-seq for each cell consists of millions of nucleotide sequences with short lengths. We first align each sequence in scRNA-seq data against reference genome and transcriptome databases. Then, to interpret various biological information in the scRNA-seq data, we parse alignment files, and build, qualify, and visualize biologically-meaningful features. To explore different aspects of RNA biology, we need to devise, implement and test various kinds of customized data analysis processes.” The data sets involved are very large, which means that parsing alignment files as part of this trial and error approach takes a lot of time. In order to shorten the timeline to develop and test data analysis codes, the team chose Julia and BioJulia.
“Julia and BioJulia are very fast and easy to write, partly due to the dynamic type system. This combination of high speed with high productivity makes Julia and BioJulia ideally suited to this computationally expensive analysis,” Dr. Ozaki explained.
Furthermore, Kenta Sato (@bicycle1885) is one of the developers of BioJulia and an intern at the Laboratory for Bioinformatics Research at the RIKEN Center for Biosystems Dynamics Research where the research was conducted. According to and Dr. Ozaki, “Kenta Sato encouraged and supported our lab members using Julia and BioJulia.”
Finally, Dr. Ozaki note: “The shell mode in Julia REPL is very useful to check input and
output files when writing codes. What’s more, the BioJulia community is very active and the developers continuously improve the BioJulia packages.” 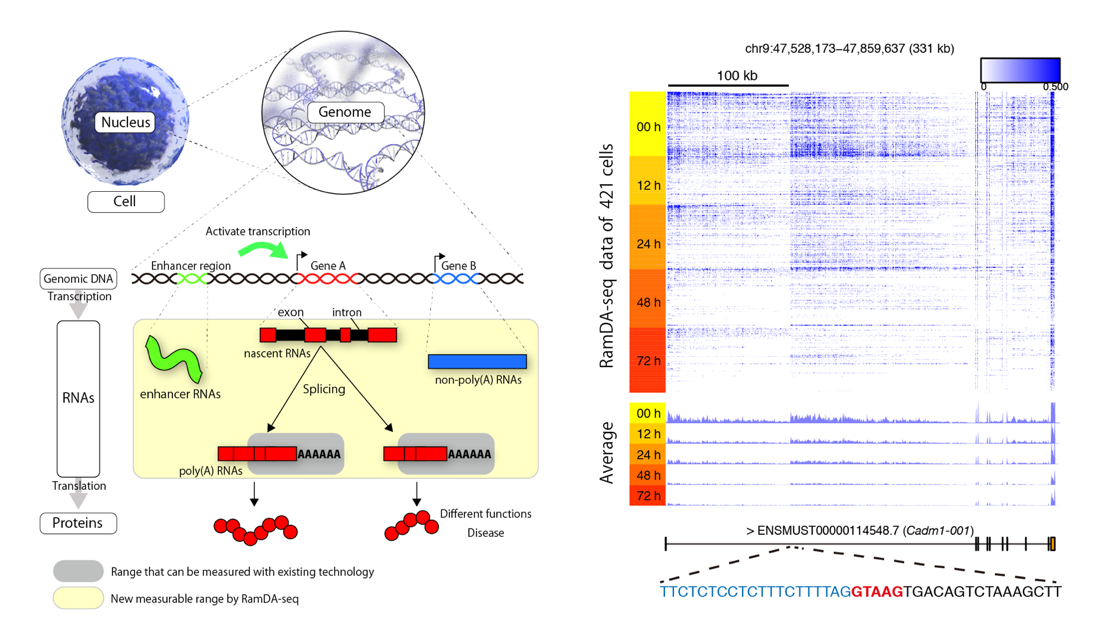
About the Author

JuliaHub
Discover key content authored by JuliaHub experts. Stay updated on innovative practices in scientific and technical computing.
